Fibrosis is a disease that is characterized by an accelerated process of collagen production and the proliferation of connective tissue in any organ of the body due to inflammation. The disease leads to tissue compaction and scar formation. When fibrosis of a particular organ develops, its functionality can deteriorate significantly. As a result, this disease leads to the development of all sorts of pathologies.
- Reasons for appearance
- Types of disease
- Symptoms of the disease
- Diagnosis and treatment
The most common fibrosis occurs in the breast and liver, lungs and prostate gland. As a result of the replacement of organ cells with connective ones, tissue elasticity decreases. In general, fibrosis is a specific reaction that tries to isolate the inflamed area from healthy tissue.
Causes and risk factors
To date, the exact causes of the development of interstitial lung diseases are unknown, but a number of predisposing factors have been identified:
- smoking;
- exposure to ionizing radiation;
- therapy with toxic drugs (bleomycin, penicillamine, cyclophosphamide);
- regular inhalation of mercury vapor, organic dust or inorganic substances;
- respiratory tuberculosis;
- recurrent viral, fungal and bacterial pneumonia;
- bronchoalveolar cancer;
- respiratory distress syndrome;
- hereditary predisposition;
- blood diseases (chronic lymphocytic leukemia, thrombocytopenic purpura, hemolytic anemia);
- collagenoses.
Forms of the disease
Interstitial lung diseases include over 130 different diseases. In 2002, the European Respiratory Society and the American Thoracic Society developed a classification system for these diseases. In accordance with it, they are divided into the following forms:
- Interstitial lung diseases of established etiology.
- Interstitial idiopathic pneumonia (cryptogenic, lymphoid, acute, desquamative, nonspecific).
- Granulomatous ILD (develops against the background of allergic exogenous alveolitis, pulmonary hemosiderosis, Wegener's granulomatosis, sarcoidosis).
- ILD associated with other diseases (malignant neoplasms, neurofibromatosis, chronic renal failure, Crohn's disease, ulcerative colitis, biliary cirrhosis, chronic hepatitis).
- Other IZL. These include forms of the disease that develop against the background of primary pulmonary amyloidosis, pulmonary proteinosis, lymphangioleiomyomatosis, histiocytosis X.
Classification of interstitial lung diseases
Interstitial lung diseases with an established etiology are divided, in turn, into the following groups:
- toxic, radiation and medicinal;
- pneumomycosis associated with HIV infection;
- ILDs arising from collagenosis (systemic lupus erythematosus, rheumatoid arthritis, dermatomyositis, scleroderma) or pneumoconiosis (berylliosis, silicosis, asbestosis);
- ILD that developed against the background of long-term infectious processes (Pneumocystis pneumonia, respiratory tuberculosis, atypical pneumonia);
- ILD due to allergic exogenous alveolitis.
How to treat pathology
In order to determine how to treat this pathology, it is necessary to establish at what stage the disease is located. Usually, at the beginning of the development of the disease, treatment is limited to various non-drug methods. Thus, breathing exercises are used for fibrosis, as well as oxygen therapy. The latter is carried out using special devices.
This allows:
- Improve the general condition of the patient;
- Eliminate shortness of breath;
- Increase performance.
Important! Such breathing exercises are impossible if there is a cough when inhaling, as well as painful sensations.
Drug treatment
If the doctor has chosen drug treatment, then in this case, glucocorticosteroids, cytostatics, and antifibrotic drugs are prescribed. This therapy lasts quite a long time (up to six months). However, regular use of such medications can negatively affect other organs of the patient. For example, if you take a full course of glucocorticosteroids, this can cause problems with blood pressure.
Surgery
If the disease has been advanced, then only surgical treatment is prescribed.
In this case, the root of the problem is eliminated by either removing the affected lung or transplanting a new one into the patient. The need for this arises if:
- The level of diffusivity decreases by more than several times;
- The development of hypoxia begins;
- Cystic fibrosis appears and cysts affect both lungs;
- The volume of the respiratory organ decreases.
Stages of the disease
In the clinical picture of interstitial lung diseases, three successive stages are distinguished:
- Spicy. The inflammatory process affects the alveolar epithelium and the capillary network of the lungs. The development of intraalveolar edema is observed.
- Chronic. There is extensive replacement of the interstitial tissue of the lungs with fibrous tissue (extensive fibrosis).
- Terminal. Fibrous tissue almost completely replaces the capillaries and alveoli, as a result of which the lung takes on the appearance of a honeycomb (the presence of multiple dilated cavities in the tissue).
The prognosis for interstitial lung disease is determined by the form of the disease. For example, patients with respiratory bronchiolitis live more than 10 years, and with Hamman-Rich syndrome no more than one year.
Classification of pulmonary fibrosis
Fibrosis is classified according to several criteria. According to the nature of the spread of the disease, it is:
unilateral, that is, only one lung is affected;- bilateral, affecting both lungs;
- focal (only a small area of the organ changes);
- diffuse (the entire lung is affected).
According to the reasons for the formation of pathology, interstitial and idiopathic fibrosis are distinguished. The idiopathic form has an unknown cause of development. It is the most diagnosed among other types of fibrosis. Most often it occurs in men 50-60 years old. The exact reasons for the development of this pathology have not been identified, but scientists have proven that it can arise due to the negative influence of genetic and environmental factors.
Interstitial fibrosis is a disease caused by exposure to negative factors.
It in turn is divided into:
- post-radiation pulmonary fibrosis, which occurs as a consequence of radiation therapy;
- dust, resulting from inhalation of dust;
- connective tissue fibrosis, the causes of which are connective tissue pathologies;
- infectious, which is a complication of infectious diseases;
- medicinal, resulting from long-term use of medications;
- peribronchial fibrosis, which is a consequence of chronic inflammation;
Fibrosis is also classified according to the severity of connective tissue formation. This classification includes:
Pneumofibrosis. It is a growth of moderate fibrous tissue that alternates with normal tissue.- Pneumosclerosis. With pneumosclerosis, there is a gross replacement of good tissue with altered tissue and compaction of the lungs.
- Cirrhosis of the lungs. Characterized by complete replacement of lung tissue and damage to the bronchi and pulmonary vessels.
Based on localization, pulmonary fibrosis is divided into:
- Apical, which affects the upper part of the organ.
- Hilar fibrosis, which affects areas near the roots of the lungs.
- Root fibrosis, which occurs at the root of the lung.
Symptoms
The clinical course of all forms of interstitial lung diseases is characterized by respiratory and general symptoms. In most cases, the disease begins gradually.
The general symptoms of ILD are vague and nonspecific. It includes:
- increased fatigue;
- general malaise;
- low-grade fever (increased body temperature up to 37.9 °C);
- loss of body weight.
Increased fatigue and general weakness are some of the symptoms of interstitial lung diseases
The first respiratory symptom of interstitial lung disease is shortness of breath. At first it occurs only under the influence of physical activity and disappears during rest, but over time it becomes permanent. Dyspnea is inspiratory in nature (it is difficult to inhale) and is often combined with wheezing.
Somewhat later, shortness of breath is accompanied by a dry or unproductive cough with scanty mucous sputum. As respiratory failure increases, cyanosis appears and increases in patients. The fingers gradually become deformed like the fingers of Hippocrates (symptoms of drumsticks and watch glasses). In some cases, deformation of the chest occurs.
In the terminal stage of the disease, symptoms of chronic pulmonary and heart failure rapidly increase in patients.
Diagnostics
With this pathology, physical changes in the lungs do not correspond to the severity of tachypnea and shortness of breath. During auscultation, the following changes are noteworthy:
- tachycardia;
- muffled heart sounds;
- crepitus during inspiration.
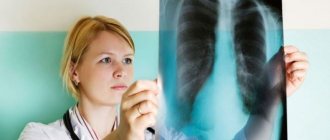
ILD is diagnosed using bronchoscopy
If interstitial lung diseases are suspected, a laboratory and instrumental examination is performed, including:
- general blood analysis;
- analysis of gas and acid-base composition of blood;
- chest x-ray;
- computed tomography of the chest;
- electrocardiography;
- diagnostic bronchoscopy;
- transbronchial, transthoracic or open lung biopsy.
Among the total number of pulmonary patients, patients with ILD make up 10-15%.
Forecast
Idiopathic pulmonary fibrosis predicts a poor prognosis. In terms of life expectancy for idiopathic pulmonary fibrosis, the estimated median survival is 2-5 years from diagnosis. Mortality rates are estimated to be 64.3 deaths per million for men and 58.4 deaths per million for women.
Mortality among patients with IPF increases with age, is higher in men than in women, and varies seasonally, with the highest mortality rates occurring in winter, even when infectious causes have been excluded.
The most common causes of death in patients with IPF include acute exacerbations of idiopathic pulmonary fibrosis, acute coronary syndromes, congestive heart failure, lung cancer, infectious causes, and venous thromboembolic disease.
Treatment
Therapy of interstitial lung diseases begins with the exclusion of further contact of the patient with provoking factors (with cessation of smoking, withdrawal of toxic drugs, cessation of interaction with harmful production factors).
To reduce the activity of the inflammatory process, corticosteroids are prescribed in high doses. As the condition improves, the dosage is gradually reduced to maintenance. If corticosteroid therapy does not lead to a lasting therapeutic effect, cytostatics are prescribed.
Corticosteroids, anticoagulants, bronchodilators, and cardiac glycosides are used in the treatment of ILD.
Also, the treatment regimen for interstitial lung diseases usually includes:
- bronchodilators;
- anticoagulants;
- cardiac glycosides;
- oxygen therapy.
In the terminal stages of the disease, the only method to save the patient's life is lung transplantation.