Symptoms of amyotrophic lateral sclerosis
There are a number of ways to relieve symptoms of the disease that help maintain maximum independence for the person with ALS and manage the emotional problems of ALS. Consultations with specialists and drug therapy are aimed primarily at maintaining a high level of quality of life for the patient for as long a period as possible.
Among neurological diseases there are those that can be successfully treated. But there are also those that can only be suspended - they belong to the group of degenerative-dystrophic ones. One such disease is ALS - amyotrophic lateral sclerosis.
ALS is a serious disease with inevitable death. Therefore, to make such a diagnosis, it is necessary to conduct a lot of research.
What is taken into account when making a diagnosis:
- The presence of an affected peripheral motor neuron, confirmed by clinical data and instrumental research methods;
- The presence of an affected central motor neuron, confirmed by clinical data;
- Progressive spread of these lesions throughout all parts of the spinal cord;
- Elimination of all other diseases that can cause such symptoms.
In this case, paresis and atrophy of the muscles of those parts of the body that are innervated by the affected neurons develop:
- Damage at the level of the brain stem - all the muscles of the face, soft palate, tongue, pharynx and larynx are affected;
- If the cervical region is affected, the muscles of the neck, upper limbs, and diaphragm are affected;
- Damage in the thoracic region - the muscles of the back and anterior abdominal wall are affected;
- When the sacral region is affected, the muscles of the back and legs are affected.
Strengthened reflexes, pyramidal signs, and spasticity appear:
- With damage to the brain stem - spasm of the masticatory muscles, signs of oral automatism, spasm of the larynx, involuntary crying and laughter;
- With a lesion in the cervical thoracic and lumbar region - spasm and hyperreflexes in the corresponding limb, small muscle spasms, pyramidal signs.
A reliable diagnosis of amyotrophic lateral sclerosis is made when peripheral and central motor neurons are damaged in three parts of the spinal cord.
What instrumental methods are used to diagnose ALS:
- Electromyography - needle and stimulation, determines damage to peripheral neurons;
- For differential diagnosis and exclusion of other diseases, computed tomography or magnetic resonance imaging is performed.
Of the laboratory methods, the only specific one is genetic mapping - identifying the genes responsible for the development of amyotrophic lateral sclerosis.
Differential diagnosis must be carried out in order to exclude all such diseases:
- Various spinal amyotrophies, including age-related ones;
- Paraneoplastic syndrome in malignant tumors;
- Hormonal imbalance;
- Some infections that have a tropism for the substance of the spinal cord;
- Vascular injuries of the spinal cord;
- Chronic intoxication with heavy metals;
- Physical damage to the spinal cord.
Amyotrophic lateral sclerosis cannot be treated. The only outcome of this disease is death from acute respiratory failure.
However, treatment is still used and has two goals:
- Slowing down the progression of the pathological process, maximizing the ability to self-care;
- Achieving a better quality of life for patients.
To achieve the first goal, only one drug is used - Riluzole. It extends the life of patients with amyotrophic lateral sclerosis by three months.
In most cases, only symptomatic treatment is used:
- For seizures, one of the following drugs is prescribed - Carbamazepine, Baclofen, Tizanil;
- Pain syndrome can only be relieved with narcotic analgesics - Tramal, Morphine;
- For depression, Amitriptyline and Fluoxetine are prescribed;
- For the correction of metabolic disorders - Cortexin, Glutoxim, Thiogamma;
- B vitamins - Combilipen, Milgamma.
For posture problems and foot deformities, corsets and orthopedic shoes are prescribed. To prevent thrombosis - compression stockings or bandaging with an elastic bandage. If the swallowing process is impaired, pureed food, feeding through a nasogastric tube.
- Multiple sclerosis in women: first signs...
- Facial nerve paresis in newborns:...
- Erased dysarthria in children - what is it and...
- Symptoms of Huntington's chorea and how to treat it...
- White matter gliosis:...
- ENMG of the upper and lower extremities - what is it...
Amyotrophic lateral sclerosis, or Lou Gehrig's disease, is a rapidly progressive disease of the nervous system characterized by damage to motor neurons in the spinal cord, cortex, and brain stem. Also, the motor branches of cranial neurons (trigeminal, facial, glossopharyngeal) are involved in the pathological process.
Any form of the disease has the same onset: patients complain of increasing muscle weakness, a decrease in muscle mass and the appearance of fasciculations (muscle twitching).
The bulbar form of ALS is characterized by symptoms of damage to the cranial nerves (9, 10 and 12 pairs):
- Those who are ill have deteriorated speech and pronunciation, and it becomes difficult to move their tongue.
- Over time, the act of swallowing is disrupted, the patient constantly chokes, and food can pour out through the nose.
- Patients feel an involuntary twitching of the tongue.
- The progression of ALS is accompanied by complete atrophy of the muscles of the face and neck; patients completely lack facial expressions, they cannot open their mouths or chew food.
The cervicothoracic variant of the disease primarily affects the patient’s upper limbs, symmetrically on both sides:
- Initially, patients feel a deterioration in the functionality of their hands, it becomes harder to write, play musical instruments, and perform complex movements.
- At the same time, the arm muscles are very tense and tendon reflexes are increased.
- Over time, weakness spreads to the muscles of the forearm and shoulder, they atrophy. The upper limb resembles a hanging whip.
The lumbosacral form usually begins with a feeling of weakness in the lower extremities.
- Patients complain that it has become more difficult for them to do work while standing, walk long distances, and climb stairs.
- Over time, the foot begins to sag, the leg muscles atrophy, and patients cannot even stand on their feet.
- Pathological tendon reflexes (Babinsky) appear. Patients develop urinary and fecal incontinence.
Regardless of which variant predominates in patients at the beginning of the disease, the outcome is still the same. The disease progresses steadily, spreading to all muscles of the body, including the respiratory muscles. When the respiratory muscles fail, the patient begins to need artificial ventilation and constant care.
In my practice, I observed two patients with ALS, a man and a woman. They were distinguished by their red hair color and relatively young age (up to 40 years). Outwardly, they were very similar: there was no hint of muscles, an amicable face, and their mouth was always slightly open.
Such patients die in most cases from concomitant diseases (pneumonia, sepsis). Even with proper care, they develop bedsores (see how and how to treat bedsores), hypostatic pneumonia. Realizing the severity of their illness, patients fall into depression, apathy, and cease to be interested in the outside world and their loved ones.
Over time, the patient's psyche undergoes strong changes. The patient I observed for a year was distinguished by his capriciousness, emotional lability, aggressiveness, and lack of restraint. Conducting intellectual tests showed a decrease in his thinking, mental abilities, memory, and attention.
The main diagnostic methods include:
- MRI of the spinal cord and brain - the method is quite informative, reveals atrophy of the motor parts of the brain and degeneration of pyramidal structures;
- cerebrospinal puncture - usually reveals normal or increased protein levels;
- neurophysiological examinations - electroneurography (ENG), electromyography (EMG) and transcranial magnetic stimulation (TCMS).
- molecular genetic analysis - studies of the gene encoding Superoxide dismutase-1;
- biochemical blood test - reveals a 5-10-fold increase in creatine phosphokinase (an enzyme formed during muscle breakdown), a slight increase in liver enzymes (ALT, AST), accumulation of toxins in the blood (urea, creatinine).
What happens in ALS
Due to the fact that ALS has similar symptoms to other diseases, differential diagnosis is made:
- brain diseases: tumors of the posterior cranial fossa, multisystem atrophy, dyscirculatory encephalopathy
- muscle diseases: oculopharyngeal myodystrophy, myositis. Rossolimo-Steinert-Kurshman myotonia
- systemic diseases
- diseases of the spinal cord: lymphocytic leukemia or lymphoma, spinal cord tumors, spinal amyotrophy, syringomyelia, etc.
- peripheral nerve diseases: Personage-Turner syndrome, Isaacs neuromyotonia, multifocal motor neuropathy
- myasthenia gravis, Lambert-Eaton syndrome - diseases of the neuromuscular junction
Treatment of the disease is currently ineffective. Medicines and proper care for the patient only prolong life expectancy without ensuring a complete recovery. Symptomatic therapy includes:
- Riluzole (Rilutek) is a well-established drug in the US and UK. Its mechanism of action is to block glutamate in the brain, thereby improving the functioning of Superoxide Dismutase-1.
- RNA interference is a very promising method for treating ALS, the creators of which were awarded the Nobel Prize in Medicine. The technique is based on blocking the synthesis of pathological protein in nerve cells and preventing their subsequent death.
- Stem cell transplantation - studies have shown that transplantation of stem cells into the central nervous system prevents the death of nerve cells, restores neural connections, and improves the growth of nerve fibers.
- Muscle relaxants – eliminate muscle spasms and twitching (Baclofen, Mydocalm. Sirdalud).
- Anabolic steroids (Retabolil) – to increase muscle mass.
- Anticholinesterase drugs (Proserin, Kalimin, Pyridostigmine) - prevent the rapid destruction of acetylcholine at neuromuscular synapses.
- B vitamins (Neurorubin, Neurovitan), vitamins A, E, C - these products improve the conduction of impulses along nerve fibers.
- Broad-spectrum antibiotics (3-4 generation cephalosporins, fluoroquinolones, carbopenems) are indicated for the development of infectious complications and sepsis.
- brain diseases: tumors of the posterior cranial fossa, multisystem atrophy, dyscirculatory encephalopathy
- muscle diseases: oculopharyngeal myodystrophy, myositis, Rossolimo-Steinert-Kurshman myotonia
- systemic diseases
- diseases of the spinal cord: lymphocytic leukemia or lymphoma, spinal cord tumors, spinal amyotrophy, syringomyelia, etc.
- peripheral nerve diseases: Personage-Turner syndrome, Isaacs neuromyotonia, multifocal motor neuropathy
- myasthenia gravis, Lambert-Eaton syndrome - diseases of the neuromuscular junction
- Riluzole (Rilutek) is a well-established drug in the US and UK. Its mechanism of action is to block glutamate in the brain, thereby improving the functioning of Superoxide Dismutase-1.
- RNA interference is a very promising method for treating ALS, the creators of which were awarded the Nobel Prize in Medicine. The technique is based on blocking the synthesis of pathological protein in nerve cells and preventing their subsequent death.
- Stem cell transplantation - studies have shown that transplantation of stem cells into the central nervous system prevents the death of nerve cells, restores neural connections, and improves the growth of nerve fibers.
- Muscle relaxants - eliminate muscle spasms and twitching (Baclofen, Mydocalm, Sirdalud).
- Anabolic steroids (Retabolil) – to increase muscle mass.
- Anticholinesterase drugs (Proserin, Kalimin, Pyridostigmine) - prevent the rapid destruction of acetylcholine at neuromuscular synapses.
- B vitamins (Neurorubin, Neurovitan), vitamins A, E, C - these products improve the conduction of impulses along nerve fibers.
- Broad-spectrum antibiotics (3-4 generation cephalosporins, fluoroquinolones, carbopenems) are indicated for the development of infectious complications and sepsis.
What should the treatment be?
Unfortunately, medicine today cannot offer effective therapy against this disease. How can you overcome ALS? Treatment should be aimed primarily at slowing the progression of the pathology. For these purposes, the following activities are used:
- special massage of the limbs;
- if the respiratory muscles fail, artificial ventilation is prescribed;
- in case of development of a depressive state, tranquilizers and antidepressants are recommended. In each specific case, drugs are prescribed individually;
- joint pain is relieved by non-steroidal anti-inflammatory drugs (Finlepsin);
- Today, experts offer all patients the drug Riluzole. It has a proven effect and is an inhibitor of the release of so-called glutamic acid. Once ingested, the drug reduces neuronal damage. However, even this remedy is not able to completely cure the patient; it only slows down the course of the ALS disease;
- To facilitate the patient's movement, special devices (canes, chairs) and collars are used to completely fix the neck.
Muscle weakness and joint stiffness
What's happening? When the number of signals from motor neurons to muscles decreases, the latter are used less and less and lose mass over time. This leads to a feeling of weakness and can cause problems with balance and gait, which increases the risk of falling.
What can be done? A decrease in muscle mass cannot be stopped by physical exercise, because... the disease progresses irreversibly. However, exercise helps maintain joint flexibility and mobility, which helps maintain muscle function, balance, and body posture. To get a referral to a physical therapist who can create an appropriate exercise program, you must contact your primary care physician.
What's happening? Due to the deterioration of signal transmission from motor neurons, muscle tension or spasms develops. This leads to impaired motor activity and coordination of movements, as well as an increased risk of falls. Sudden muscle spasms can be extremely painful.
What can be done? To eliminate this symptom, as a rule, it is enough to change the position of the body while resting in a bed or chair. Exercise partially solves the problem. In addition, your doctor may prescribe medications to relax your muscles.
What help does the patient and his family need?
Photo by Yuri Mozolevsky from the site sb.by
A person with ALS may need consultations with a neurologist, respiratory support specialist, physical therapist, occupational therapist, nutritionist, speech therapist, music therapist, psychologist, palliative care physician, as well as the help of a nurse and visiting nurse.
The family will have to provide him with certain equipment. This could be a wheelchair, walker, multifunctional bed, ventilator or NIV, aspirator (saliva ejector), cougher, lift (for example, for moving from a bed to a wheelchair or from a stroller to a bath). In addition, the patient needs medicine, nutritional therapy, hygiene products, etc.
Each patient with ALS needs an individual route of care, as the disease can progress in different ways.
Characteristics of the pathology
With amyotrophic lateral sclerosis, there is a gradual death of nerve cells located in the lateral horns of the spinal cord and responsible for motor function.
At the same time, various movement disorders develop. The disease gradually progresses and ultimately leads to death due to damage to the respiratory muscles.
Who is sick?
ALS is a fairly rare disease. In the world, no more than three out of one hundred thousand people get sick with it. Under the age of 65, the disease is one and a half times more common in men, then men and women are equally affected.
Amyotrophic lateral sclerosis can occur at any age - from teenagers to old people. In ten percent of cases, this is a familial form of the disease. The remaining cases are sporadic.
Classification
There is no single cause for the development of amyotrophic lateral sclerosis; it occurs as a result of a combination of several factors.
Causal factors can come from the body itself and from the external environment:
- Genetic disorders - 18 genes found responsible for the development of ALS;
- Neurotropic viruses and prions;
- Bad ecology;
- Excessive stress.
ALS is a disease that develops in patients of working age, which determines the importance of early diagnosis. The pathology occurs with a frequency of 2-5 cases per 100 thousand population. Men get sick more often. ALS accounts for 80% of the total number of motor neuron diseases. ALS disease has no cure.
Amyotrophic lateral sclerosis, also called ALS, is a disease that in 10% of cases manifests itself as progressive bulbar palsy, which indicates damage to the nuclei that form the basis of the cranial nerves. Muscle atrophy is detected in 8% of patients and is also progressive.
Anatomy of the pyramidal tract
The anatomy of the pyramidal tract is presented here. Here you see the supplementary motor area, the premotor cortex.
Pyramidal tract
These transformations transmit signals from the brain to motor neurons in the spinal cord. They innervate skeletal muscles and regulate voluntary movements.
Amyotrophic lateral sclerosis is an unusual neurodegenerative disease! The ICD 10 code is G12.2.
All the worst things happen when a person doesn’t feel anything yet. At this preclinical stage, 50–80% of motor neurons die after a genetic failure occurs with the participation of environmental factors. Then, when 20% of resistant motor neurons remain, the disease itself begins.
Manifestation of amyotrophic lateral sclerosis
Clinical picture
Symptoms of amyotrophic lateral sclerosis vary depending on the level of damage:
- With pathology in the cervical and thoracic spinal cord, the first symptoms will be the development of flaccid paralysis of the upper limbs. In this case, an increase in all reflexes will be observed. Then signs of tense paralysis of the lower extremities appear, and all reflexes also intensify.
- Pathological signs of damage to the pyramidal tracts appear. Muscle atrophy gradually develops. The progression of the disease leads to impaired mobility of the diaphragm and intercostal muscles - as a result, acute respiratory failure develops.
- If the disease begins with damage to the lumbar and cruciate regions, the first to develop is flaccid paralysis of the lower extremities and increased reflexes. Pathological pyramidal symptoms are added.
- Then tension paralysis of the upper limbs with hyperreflexia is formed. Upper limb paralysis reaches its maximum later than lower limb paralysis. In both cases, the last stage is signs of bulbar insufficiency.
There is a bulbar form - with it the disease begins with the following symptoms:
- Speech impairment;
- The voice becomes nasal;
- Atrophy of the tongue muscles develops, as a result of which the swallowing process is disrupted;
- After this, paresis of the soft palate appears, reflexes intensify, and signs of oral automatism appear;
- Gradually, paralysis affects the upper and then lower limbs;
- In this case, enhanced reflexes and pyramidal signs are observed;
- This form of the disease is accompanied by a sharp decrease in body weight due to difficulties with eating;
- At the last stage, respiratory failure develops.
The most unfavorable variant of the development of amyotrophic lateral sclerosis is the primary generalized form:
- Paralysis of all limbs immediately develops;
- Tendon reflexes are weakened;
- Almost immediately, bulbar syndrome appears with impaired speech and swallowing;
- The patient quickly loses body weight;
- Respiratory failure develops quickly.
Natural pathomorphosis
If we talk about various debuts, it should be noted that the sequence of development of symptoms is always certain.
Pathomorphosis in bulbar and cervical onset of ALS
With bulbar onset, speech disturbances first occur. Then there are swallowing disorders. Next, paresis occurs in the limbs and respiratory disorders.
With cervical onset, the process of disruption begins with one arm and then moves to the other. After this, bulbar disorders and movement disorders in the legs may occur. It all starts on the side where the primary hand was injured.
If we talk about the thoracic onset of ALS, the first symptom that patients usually do not notice is weakness of the back muscles. The condition is broken. Then paresis occurs in the hand along with atrophy.
Pathomorphosis in thoracic and lumbar onset of ALS
Then the disease spreads to the leg of the same side. Gene paresis occurs. The legs are affected and then bulbar and respiratory disorders occur.
In lumbar onset, one leg is affected first. Then the second one is captured, after which the disease spreads to the hands. Then respiratory and bulbar disorders occur.
Fatigue
What's happening? Decreased physical muscle functionality requires greater energy expenditure to maintain daily activity. Other causes of fatigue include breathing problems, shortness of breath, decreased food intake, and dehydration.
What can be done? Make a plan to complete your tasks for the day. This will help maintain a balance between activity and adequate rest. A physical therapist can offer more detailed techniques for solving the problem of fatigue. It is also important to consult with a nutritionist about increasing calorie intake and fluid intake.
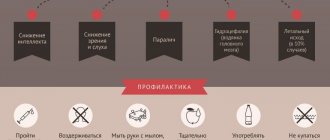
How not to start the disease
It is almost impossible to understand the development of this disease at first, since the exact causes of its occurrence are not fully understood. Secondary preventive measures are aimed at slowing the progression of the disease. These include:
- Regular consultations with a neurologist and taking medications.
- Rejection of bad habits.
- Correct and competent treatment.
- Balanced diet and vitamin intake.
ALS is an incurable disease; scientists to this day have not established its exact signs and causes. At this stage of medical development, there is no drug that could completely cure a person of an illness.
Diagnosis of amyotrophic lateral sclerosis
- MRI of the brain and spinal cord. It is carried out to exclude other pathologies of the brain substance.
- Electromyography. It is carried out to confirm the presence of a denervation process (separation of nerve connections between muscles and the nervous system) and determine the degree of damage to the brain matter.
- Biopsy of muscle tissue. Signs of muscle atrophy due to denervation are detected, often earlier than pathological changes are observed during electromyography.
To make a diagnosis, the following conditions are necessary: the presence of signs of degenerative changes in motor neurons, the spread of the pathological process, the absence of other pathologies with identical symptoms. Spirography (measurement of speed and volume parameters of breathing) and polysomnography (sleep study) are carried out to determine the degree of dysfunction of the respiratory system.
- consultation with a neurologist;
- electromyography;
- MRI, CT;
- laboratory examinations (general clinical tests, biochemistry, microscopy and culture of cerebrospinal fluid);
- PCR tests (detection of mutations in certain genes).
The doctor carefully interviews the patient, clarifying the family history. The survey determines whether any of the relatives suffered from chronic progressive movement disorders.
For reference. The diagnosis is confirmed by electromyography, which reveals rhythmic fasciculatory potentials with an amplitude of up to 300 μHz and a frequency of 5-35 Hz (ri).
MRI and CT scans should exclude all other possible diseases of the nervous system that have similar symptoms.
- clinical examination and questioning of the patient about the course of the disease and life;
- neurophysiological examination methods (EEG, evoked potentials, etc.);
- neuroimaging methods (computer and magnetic resonance imaging);
- genetic research methods to establish the fact of hereditary predisposition;
- blood tests for infections (syphilis, HIV, etc.);
- cerebrospinal fluid studies.
What happens to the patient
What's happening? ALS does not directly cause pain or discomfort. But they can be the result of a number of other reasons. For example, pain occurs as a result of muscle spasms, general spasticity, muscle tension, skin pressure, or constipation. Therefore, it is important to find out the cause of the symptom.
What can be done? There are recommendations for optimal body positions, support, prevention of local compression and drug therapy. In case of prolonged pain, you should contact a medical facility. The doctor can choose the appropriate pain reliever.
Problems with speech and communication
What's happening? When the muscles of the face, mouth and larynx are affected, swallowing becomes difficult. Disruption of the normal process of eating and swallowing is called dysphagia. As a result, a person receives less nutrients and fluids, which can lead to weight loss.
What can be done? It is necessary to contact a speech therapist and nutritionist, who will assess the degree of swallowing impairment and changes in body weight, and also talk about possible solutions to the problem. Including, in order to increase the intake of proteins and carbohydrates from food, you need to adjust your diet. There are also alternative methods that can serve as support or complete nutritional replacement.
What's happening? With ALS, the respiratory muscles are sooner or later affected. As the disease progresses—especially in the later stages—breathing problems develop. When this happens, the patient will need breathing devices and specialist consultation.
What can be done? If a person experiences shortness of breath, weakness, sleep disturbances, morning headaches, or sleepiness during the day, their healthcare provider may refer them to a pulmonologist. Methods for correcting the problem may include breathing and physical exercises, recommendations for creating a comfortable body position, effective coughing techniques, drug therapy and special equipment for ventilation.
What's happening? As the muscles of the face and larynx weaken, and ventilation of the lungs further decreases, it becomes increasingly difficult for a person to speak. This speech difficulty is called dysarthria.
What can be done? Your doctor will help you assess the problem and select techniques to solve it. We also recommend consulting with a physiotherapist, who will recommend equipment or aids depending on what manipulations a person with ALS is capable of. Tools for speech and communication (they are also called “means of alternative and auxiliary communication”) include both simple techniques (gestures, writing, alphabet tables, etc.) and more technically complex ones (using a computer).
Who helps people with ALS in Russia
Photo from the website camilliani.org
After registering a disability, a patient with ALS will be able to receive various technical equipment and consumables from the social security authorities under the IPRA (individual program for rehabilitation and habilitation): a wheelchair, walkers, and hygiene products. But this is a lengthy process that requires collecting numerous certificates.
Systemic support for patients and their families is provided by the Live Now Charitable Foundation. The service for helping people with ALS, the main project of the foundation, provides consultations with doctors, assistance from substitute caregivers, and trains relatives to care for patients.
If necessary, the fund provides patients with breathing equipment and consumables. Ventilators and NIV devices, as well as coughers, are provided on a free rental basis.
Every month, the ALS service runs outpatient clinics, or “clinics,” where people with ALS can see all the key specialists in the disease, receive advice, and see other patients in one day and in one place.
One of the important areas that the Live Now Charitable Foundation plans to develop is the preparation of educational programs for doctors in the regions.
In addition, the foundation has a Family Council, which organizes various events for communication and recreation for patients. “Communication is very important for ALS patients. They are often embarrassed by their illness and become despondent,” the foundation noted.
Useful addresses and telephone numbers can be found in the link
Epidemiology of the disease
The disease is extremely rare, approximately 2-5 people per 100,000. It is believed that men over 50 years of age are more often affected. Lou Gehrig's disease makes no exceptions for anyone; it affects people of different social status and various professions (actors, senators, Nobel laureates, engineers, teachers). The most famous patient was world baseball champion Loi Gering, after whom the disease took its name.
In Russia, amyotrophic lateral sclerosis has become widespread. Currently, the number of sick people is approximately 15,000-20,000 in the population. Among the famous people in Russia who have this pathology are composer Dmitry Shostakovich, politician Yuri Gladkov, and pop performer Vladimir Migulya.
The causes of ALS are not entirely known; it is believed that it is caused by exposure to a number of external factors (toxic substances or undetected microorganisms). All of them contribute to the death of neurons responsible for motor movements, which leads to the appearance of characteristic symptoms. Although the causes of ALS are not fully known, however, scientific work is underway to identify them and seek to prevent the disease.
Development of the disease and characteristic signs of amyotrophic lateral sclerosis. Find out how the disease progresses.
You can find out everything about diagnosing amyotrophic lateral sclerosis using neuroimaging methods here.
Pathogenetic therapy for Lou Gehrig's disease
Here we see many types of familial ALS. More than 20 mutations have been discovered. Some of them are rare. Some are common.
| Type | Frequency | Gene | Clinic |
| FALS1 (21q21) | 15-20% FALS | SOD-1 | Typical |
| FALS2 (2q33) | Rare, AP | Alsin | Atypical, SE |
| FALS3 (18q21) | One family | Unknown | Typical |
| FALS4 (9q34) | Very rare | Sentaxin | Atypical, SE |
| FALS5 (15q15) | Rare, AR | Unknown | Atypical, SE |
| FALS6 (16q12) | 3-5% FALS | FUS | Typical |
| FALS7 (20p13) | One family | ? | Typical |
| FALS8 | Very rare | VAPB | Atypical, different. |
| FALS9 (14q11) | Rare | Angiogenine | Typical |
| FALS10 (1p36) ALS-FTD | 1-3% Fals up to 38% familial and 7% sporadic | TDP-43 9q21, C9orf72 | Typical ALS, FTD, ALS-FTD |
There are also new ALS genes. We didn't show some of them. But the point is that all these mutations lead to one final path. To the development of damage to central and peripheral motor neurons.
There are medications that slow the progression of Lou Gehrig's disease.
Riluzole is a presynaptic inhibitor of glutamate release. Extends the life of patients by an average of 3 months. It must be taken as long as the person maintains self-care. Doses of 50 mg 2 times a day before meals every 12 hours.
Riluzole (Rilutek)
In 3 - 12% of cases, the drug causes drug-induced hepatitis and increased blood pressure. Metabolized in men and smokers. They require a higher dose.
It is worth saying that a slowdown in progression cannot be felt. The drug does not make a person better. But the patient will be sick longer and later stops caring for himself.
The drug is contraindicated in patients with definite and probable ALS with a disease duration of less than five years with a forced vital capacity of more than 60% and without tracheostomy.
The price of Riluzole in different pharmacies ranges from 9,000 to 13,000 rubles.
NP001 - active substance sodium chlorite. This medicine is an immune regulator for neurodegenerative diseases. Suppresses macrophage inflammation in vitro and in patients with amyotrophic lateral sclerosis.
Sodium chlorite
3 months after droppers at a dose of 2 mg/kg, sodium chlorite stabilizes the course of the disease. It seems to stop the progression.
Saliva and phlegm
What's happening? When swallowing is impaired, excess saliva accumulates in the oral cavity, which leads to drooling and the associated feeling of discomfort. The consistency of the secretion can be either watery or thick. Increased viscosity is associated with a decrease in the amount of fluid entering the body.
What can be done? Options for solving this problem include dietary adjustments, drug therapy, and the use of aspiration devices for cleaning the oral cavity (suction).
Symptoms of ALS
As a rule, the disease is diagnosed in adulthood (after 40 years), and the risk of getting sick does not depend on gender, age, ethnic group or other factors. Sometimes there are cases of a juvenile form of pathology, which is observed in young people. In the first stages of ALS, there are no symptoms, after which the patient begins to experience mild cramps, numbness, twitching and muscle weakness.
The pathology can affect any part of the body, but usually (in 75% of cases) it starts from the lower extremities - the patient feels weakness in the ankle joint, which is why he begins to stumble when walking. If symptoms begin in the upper extremities, the person loses flexibility and strength in the hands and fingers. The limb becomes thinner, the muscles begin to atrophy, and the hand becomes like a bird's paw. One of the characteristic signs of ALS is asymmetrical manifestations, that is, symptoms first develop on one side of the body, and after some time on the other .
In addition, the disease can occur in the bulbar form - affecting the speech apparatus, after which difficulties arise with swallowing function, and severe salivation appears. The muscles responsible for chewing function and facial expressions are affected later, as a result of which the patient loses facial expressions - he is unable to puff out his cheeks, move his lips, and sometimes stops holding his head up normally. Gradually, the pathological process spreads to the entire body, complete muscle paresis and immobilization occur. There is virtually no pain in people diagnosed with ALS; in some cases, it occurs at night and is associated with poor mobility and high spasticity of the joints.
Table. Main forms of pathology.
| Form of the disease | Frequency | Manifestations |
| Cervicothoracic | 50% of cases | Atrophic paralysis of the upper and lower extremities, accompanied by spasms |
| Bulbarnaya | 25% of cases | Paresis of the palatine muscles and tongue, speech disorders, weakening of the masticatory muscles, after which the pathological process affects the limbs |
| Lumbosacral | 20-25% of cases | Signs of atrophy are observed with virtually no disturbance in the tone of the leg muscles; the face and neck are affected in the last stages of the disease |
| High | 1-2% | Patients experience paresis of two or all four limbs, unnatural expression of emotions (crying, laughter) due to damage to the facial muscles |
The above signs can be called average, since all patients with ALS manifest themselves individually, so it is quite difficult to identify specific symptoms. Early symptoms may be invisible both to the person himself and to others - there is slight clumsiness, awkwardness and slurring of speech, which is usually attributed to other reasons.
Important: cognitive functions are practically not affected in ALS - moderate memory impairment and impairment of mental abilities are observed in half of the cases, but this makes the general condition of patients worsen even more. Due to the awareness of their own situation and the expectation of death, they develop severe depression.
Causes of amyotrophic lateral sclerosis
The causes of amyotrophic lateral sclerosis are not fully understood. Pathogenetic mechanisms are constantly being studied, which is associated with the emergence of many theories and hypotheses:
- Glutamate excitotoxicity. The pathogenesis is based on malfunctions of the glutamate-aspartate system, which provides the transport function. As a result of disturbances, excitatory acids accumulate in the motor areas of the central nervous system.
- Autoimmune reaction. The pathogenesis is based on the production of antibodies to calcium channels, which triggers a series of reactions leading to the death of motor neurons.
- Deficiency of neurotrophic factor - proteins that stimulate and support the development of neurons.
- Mitochondrial dysfunction. The pathogenesis is based on increased permeability of mitochondrial membranes.
Neither theory is supported by conclusive evidence. In 90% of cases, the disease develops spontaneously, sporadically. In 10% of cases, the causes of ALS correlate with hereditary predisposition. In patients with a hereditary form, a mutation of the SOD-1 enzyme (superoxide dismutase enzyme) is detected in 20% of cases.
The SOD-1 enzyme is involved in regulating the amount of free radicals. Amyotrophic lateral sclerosis is a disease, the development of which in most cases occurs spontaneously, but under the influence of favorable conditions, which allows us to identify risk factors:
- Age over 50 years.
- Hereditary predisposition.
- Male gender.
- Bad habits (smoking, alcoholism).
- Residence in rural areas (correlation with negative health effects of pesticides used in agriculture).
Amyotrophic lateral sclerosis syndrome is characterized by clinical heterogeneity (heterogeneity), which leads to many options for describing the clinical picture in practical neurology. Heterogeneity is manifested in the localization of the onset pathological focus, the nature of the spread of the pathological process, the variability of the combination of symptoms, and the rate of progression of neurological deficit.
The disease is based on the accumulation of pathological insoluble protein in the motor cells of the nervous system, leading to their death. The cause of the disease is currently unknown, but many theories exist. The main theories include:
- Viral - this theory was popular in the 60-70s of the 20th century, but was never confirmed. Scientists in the USA and USSR conducted experiments on monkeys, injecting them with extracts of the spinal cord of sick people. Other researchers tried to prove the participation of the polio virus in the formation of the disease.
- Hereditary - in 10% of cases the pathology is hereditary;
- Autoimmune - this theory is based on the discovery of specific antibodies that kill motor nerve cells. There are studies that prove the formation of such antibodies against the background of other serious diseases (for example, lung cancer or Hodgkin's lymphoma);
- Genetic - 20% of patients have disorders of the genes encoding the very important enzyme Superoxide dismutase-1, which converts Superoxide, which is toxic to nerve cells, into oxygen;
- Neuronal - British scientists believe that glial elements, that is, cells that ensure the vital activity of neurons, are involved in the development of the disease. Studies have shown that if astrocytes, which remove glutamate from nerve endings, have insufficient function, the likelihood of developing Lou Gehrig's disease increases tenfold.
Amyotrophic lateral sclerosis occurs with a frequency of 1-5 cases per 100 thousand population. The pathology often debuts at the age of 50-70, but cases of morbidity are also recorded at a younger age. Men get ALS more often than women.
Risk factors for the development of amyotrophic lateral sclerosis are:
- Smoking.
- Age over 50 years.
- Male gender.
- Intense physical labor.
- History of trauma.
- Infectious diseases.
- Previous surgical interventions.
- Hypovitaminosis.
For reference. There is no single reason why pathology occurs. Amyotrophic lateral sclerosis is considered to be a multifactorial disease. A combination of factors takes part in its development, the leading of which is recognized to be genetic.
Since 1990, more than 20 genes have been identified, mutations and damage to which can lead to the development of ALS. The difficulties in studying the genetic mechanism of the development of the disease are as follows:
- in most patients, the pathology manifests itself at a late age, and not all people live to see its manifestations: hereditary predisposition cannot always be reliably tracked, patients may not know that their relative was a carrier of the ALS genes;
- Carriage of mutated genes does not mean the appearance of the disease; carriers have a less than 50% chance of getting the disease
A mutation in the superoxide dismutase 1 (SOD-1) gene can lead to:
- degenerative changes in motor neurons due to the destruction of mitochondria,
- interruption of axonal transport,
- accumulation of metabolic products harmful to the cell,
- accumulation of excess intracellular calcium,
- increased load on motor neurons,
- damage to the surrounding microglia.
In the course of ongoing clinical trials, funded from funds raised through a flash mob in 2014, scientists from the global Project MinE identified a new gene, NEK1, “culpable” for the occurrence of the disease. This discovery can rightfully be considered a scientific breakthrough in the study of ALS.
By frequency of occurrence:
- Classic ALS
- Progressive bulbar palsy
- Progressive muscular atrophy
- Primary lateral sclerosis
- Western Pacific complex (ALS-parkinsonism-dementia)
| By frequency of occurrence: | By inheritance: |
|
|
| According to the level of damage to the central nervous system: | Nosological forms of the disease: |
|
|
Causes and risk factors
As a nervous disease, amyotrophic lateral sclerosis has been known for a long time, but the causes of the disease are still largely unknown. The disease occurs both as a result of hereditary predisposition and spontaneously. Most cases still occur spontaneously.
One in 100 ALS sufferers has an inherited or reoccurring mutation in a specific gene that is important for cellular metabolism.
This prevents the formation of an enzyme that removes oxygen free radicals in the cell. Unremoved radicals can cause significant damage to nerve cells, interfering with the transmission of commands to muscles - resulting in muscle weakness and, ultimately, complete paralysis.
Emotional lability (pseudo-bulbar effect)
What's happening? Some people with ALS have bouts of uncontrollable laughter and/or crying that are difficult to control. These reactions do not occur in all patients, and they are involuntary.
What can be done? To relieve symptoms, you can turn to drug therapy. These reactions may cause some concern for others, but if they know that these manifestations are part of the symptoms of ALS, it will be easier for them to cope with it.
What's happening? Some people with ALS experience a range of emotional states, including anxiety, fear, anger, sadness, depression and denial. These reactions are normal.
What can be done? Awareness of your emotional states is the first step to solving problems associated with feelings. If these conditions are too pronounced and persist for a long time, we strongly recommend that you seek help from a doctor. In some cases, drug therapy and/or psychotherapy are effective.