Синдром Марфана – наследственная патология, в результате прогрессирования которой происходит поражение соединительной ткани. В процесс также вовлекается скелетно-мышечная система и зрительный аппарат. Ткань внутренней среды (соединительная) выполняет массу важных функций, одной из которых является соединение определённых частей тела, и закладывание основы для их нормального роста и развития. В случае прогрессирования синдрома Марфана, в структуре ткани появляются дефекты, которые не дают ей функционировать так, как необходимо.
- Причины
- Классификация
- Симптоматика
- Скелет
- Зрение
- Сердце и сосуды
- Нервная система
- Кожный покров
- Лёгкие
- Диагностика
- Лечение
Так как данная ткань располагается по всему телу, синдром Марфана оказывает влияние и на остальные органы и системы:
- скелет;
- сердце;
- сосуды;
- зрительный аппарат;
- кожный покров;
- лёгкие;
- ЦНС.
Синдром Марфана в одинаковой мере поражает и мужчин, и женщин. Патологию диагностируют у людей всех этнических групп и рас. Распространённость – 1 случай на 10 тысяч человек. Риск рождения ребёнка с данной болезнью повышается в несколько раз в том случае, если отец достигает возраста 35 лет. В 50% случаев ребёнок рождается с патологией, если у одного из родителей диагностирован синдром Марфана. Тип наследования – аутосомно-доминантный.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Дифференциальная диагностика
Проводят дифференциальную диагностику с заболеваниями, имеющими схожую клиническую картину.
- Синдром Билса. Данная болезнь (врождённая контрактурная арахнодактилия), характеризуется наличием дефекта в гене FBN2, кодирующий белок фибрилин 2. Клинические проявления при таком состоянии практически не отличаются от синдрома Марфана. Единственным отличием служит отсутствие поражения зрения при синдроме Билса. И на первое место выходит изменение пальцев рук с развитием контрактур (невозможность движений).
- Гомоцистинурия — наследственное заболевание, в основе которого лежит нарушение обмена серосодержащих аминокислот. Диагноз можно поставить уже в раннем возрасте, опираясь на клинические проявления. Помимо схожих с синдромом Марфана симптомов, наблюдаются: отставание в физическом и нервно-психическом развитии, умственная отсталость, судороги, гиперкинезы (размашистые движения), поведенческие нарушения. Внешний вид очень схож с синдромом Марфана, но определяющая роль принадлежит умственной отсталости, которая наблюдается при гомоцистинурии.
- Синдром Элерса — Данлоса, который характеризуется гиперэластичностью кожи. Развивается при нарушении синтеза белка коллагена. Наряду со схожими симптомами синдрома Марфана, у пациента на первое место выходит поражение кожного покрова. Кожа имеет высокую эластичность и растяжимость, а также хрупкость и ранимость. Незначительные микротравмы и порезы длительно заживают и способны оставлять после себя рубцы.
- Синдром Лойса — Дитца. Такой синдром проявляется при дефекте в генах, отвечающих за синтез транформирующего фактора роста бета-1. Отличительной особенностью данного заболевания является гипертелоризм (широко посаженные глаза), порок развития лицевого скелета «волчья пасть» (расщепление твёрдого нёба).
Симптомы синдрома Марфана
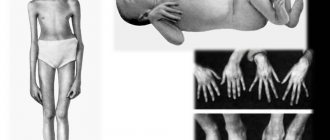
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов; — аномалии строения тазобедренного сустава; — кифоз, сколиоз; — вывихи шейного сегмента позвоночника; — деформация грудной клетки; — плоскостопие; — глубокая посадка глаз; — уменьшенная нижняя челюсть, нарушение роста зубов; — высокое нёбо; — атрофические «растяжки» на коже; — паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения); — пороки сердца (чаще — поражения клапанов); — стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
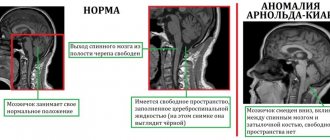
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Что это за болезнь?
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения: — прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.); — хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Причины
Основная причина прогрессирования синдрома Марфана – мутация гена FBN1. Он отвечает за синтез фибриллина. Данный белок выполняет чрезвычайно важную функцию в организме – он отвечает за сократимость и эластичность соединительной ткани.
Нарушение формирования волокон соединительной ткани и утрата ими прочности, происходит ещё во время внутриутробного развития плода. Это приводит к тому, что ткань перестаёт выдерживать естественные нагрузки. Атипичные изменения наблюдаются в крупных сосудах мышцах, клапанах сердца. Если не проводить адекватную терапию, то продолжительность жизни человека, больного синдромом Марфана, составляет всего 40 лет. Правильно подобранный план лечения даёт возможность увеличить эту цифру в два раза.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:
Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Лечение патологии
Дети, у которых диагностирован синдром Марфана следует проходить правильное лечение, которое предполагает прием большого количества препаратов. Их действие направлено на стабилизацию работы сердечно-сосудистой системы, стимуляцию центральной нервной системы. Также рекомендуется назначение энерготропных препаратов и антиоксидантов. При этом врач сочетает их прием с проведением целого ряда терапевтических мер, которые необходимы для лечения данного заболевания.
Процесс лечения должен включать прием бета-адреноблокаторов таких, как Обзидан, Атенолол по 10 миллиграмм в сутки. При этом длительность приема от 6 до 12 и более месяцев. Распространено применение энерготропных и антиоксидантных препаратов: Рибоксин, Витамины В1 В2, аскорбиновая кислота, Токоферол, Элькар, Димефосфон, Лимонтар, ноотропные препараты, Пирацетам.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
- аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
- выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Почему проявляется генетический синдром?
Причиной развития недуга считается мутация в гене FBN1, который располагается в 15 хромосоме и отвечает за нормальное производство фибриллина 1. Этот белков соединительной ткани является одним из главных компонентов, придающих ей эластичность и способность к сокращению.
Первыми при генетическом синдроме поражаются структуры, содержащие наибольшее количество важного белка – стенки кровеносных сосудов, связочный аппарат, цинновая связка глаза. Изменённая соединительная ткань не способна выполнять своей функции, выдерживать физическую нагрузку в связи с потерей прочности и упругости, у ребёнка возникают симптомы заболевания.
Недуг относится к генетическим и передаётся от родителей по аутосомно-доминантному типу. Риск появления малыша с наследственным синдромом очень высокий, если у мамы или папы имеются признаки болезни. В 75% случаях заболеваний прослеживается появление недуга в каждом поколении семьи. У 25% больных определяется новая, спонтанная мутация, не находится чёткой связи с наследованием.
Соединительная ткань не образует отдельного органа в человеческом теле. Но её клетки располагаются во всём организме. По средствам этих структур выполняются опорная, защитная и трофическая функции, образуется своеобразный каркас и покровы всех органов. К разновидностям соединительной ткани относят хрящевую, костную, мышечную, жировую ткани, кровь и лимфу. Поэтому системные заболевания, связанные с тканевой патологией, отличаются большим многообразием проявлений.